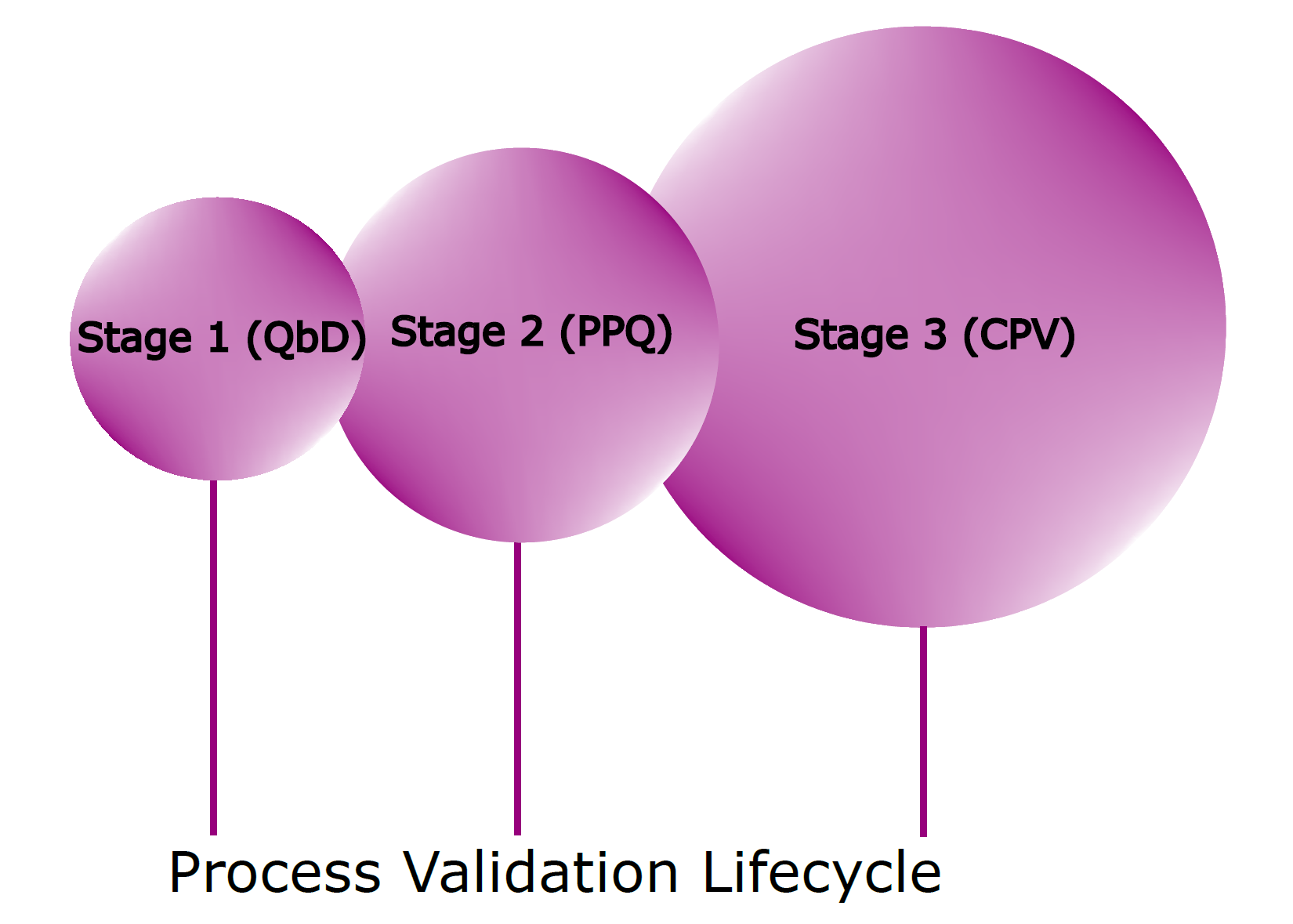
Almost a decade old, the FDA’s 2011 Process Validation Guidance is a fundamental shift from the process validation approach previously followed for decades. It stipulates process validation activities in three different stages: Stage 1 – process design, Stage 2 – process qualification, and Stage 3 – continued process verification (CPV). 1 Understanding and using the growing body of process knowledge gained through the process validation life cycle is the current expectation. The stages are interconnected such that data from all stages are required to be used to make informed science- and data-driven decisions. Process validation is no longer a singular finite activity; it is a continuous activity starting with Stage 1, quality by design (QbD) process development, covering Stage 2, and continuing through to Stage 3, which may include heightened sampling and testing while under routine monitoring. 2 As changes are introduced, the cycle goes back to Stage 1 for evaluation.
Continuous Process Verification
The third phase of the life cycle, continued process verification, is the ongoing assurance that the routine process remains in a state of control. When product knowledge and process understanding are gained through Stage 3, the expectation is to use that information immediately for continuous improvement actions. A false perception that the annual product quality review (APQR) replaces Stage 3 is still prevalent across the industry, and it should be recognized that, unlike the APQR, the CPV program requires a system for detecting drifts promptly. However, the ongoing Stage 3 data and assessments can be used to complete the applicable APQR report sections, thus avoiding duplication.
Stage 3 of the life cycle typically garners the most process data, allowing for the enhancement of the control strategy on an ongoing basis. The continued or ongoing process verification stage is split into two stages – 3a and 3b. While the guidance does not delineate Stage 3a and Stage 3b, there is a need to distinguish these two phases for better understanding and control of the process. A Stage 3a assessment speaks specifically to understanding and managing process variability of your new process. The protocol-based Stage 3a assessment provides the basis for moving into a standard operating procedure (SOP)-driven ongoing Stage 3b plan. With an effective Stage 3, the need for the antiquated revalidation requirement is negated.
Figure 1: Stage 3, a vital source of process data
Since Stage 3 (CPV) never ends, a preliminary assessment with a substantial body of data (Stage 3a) is recommended before routine monitoring. Stage 3a presents an opportunity to statistically analyze Stage 1, Stage 2, and Stage 3 all at once, allowing for in-depth process understanding. It includes analyzing the impact of material attributes and process parameters and their impact on quality attributes, an assessment that was performed during the Stage 1 QbD development phase. At Stage 3a, either the QbD design space is reconfirmed or shifts in process are observed. If a misalignment with the original design space estimates is observed, this may call for continuous improvement or further process optimization.
Understanding Process Variability
Stage 3a is a critical tool for understanding inherent process variability. The sources of critical quality attribute (CQA) variability can be method, material, mother nature (environmental conditions), man, machine, and measurement. If the Stage 3a phase can have minimal planned input changes, it may allow for control of the majority of output variables, which can then lead to determining the true process and analytical variability. Variability within batches indicates analytical (measurement) causes, variability between batches indicates process-related issues. Deducing analytical and process variability, the major contributing factors, is key insight for implementing continuous improvement strategies. Once the process variability measure is understood, it can be used to calculate business operational indices such as the PaCS Index, 3 which is applied to make business decisions such as manufacturing process continuity, manufacturing site preference, etc. Correlation of bio-clinical in-vivo results with Stage 3a in-vitro dissolution profiles, where possible, may further confirm the product’s consistent dissolution performance. Organizations can use successful completion of Stage 3a as a research and development (R&D) to commercial handover criteria.
Using Process Signals During Routine Monitoring
When moving into Stage 3b routine monitoring post Stage 3a, it is critical to understand the process signals generated from Stage 3b. The signals representing failure will be addressed by the site quality management system and investigated according to standard operating procedures. The remainder that are out of statistical control are not failures but are yellow flags indicating process variability and drifts. The guidance is very specific in stating that trending should be performed in such a way so as to guard against overreaction to individual events, so automatically categorizing them as quality failure investigations without additional review of the signals is not an ideal approach. Industry groups are developing guidelines on how to address common Stage 3 (CPV) signals.
In addition to serving as a continuous improvement tool and helping to meet regulatory guidance expectations, Stage 3b data can be used for business purposes. For example, blend scale-up process parameters for a product may be determined based on blend results and process parameters of a similar formulation, with similar material properties, same bin fill, etc. The use of similar product/process data, as allowed in the process validation guidance, can in fact reduce the need for unnecessary trials and help move into scale-up experimental batch manufacture quickly. If Stage 3b data is readily available for use, its application is limitless. 4
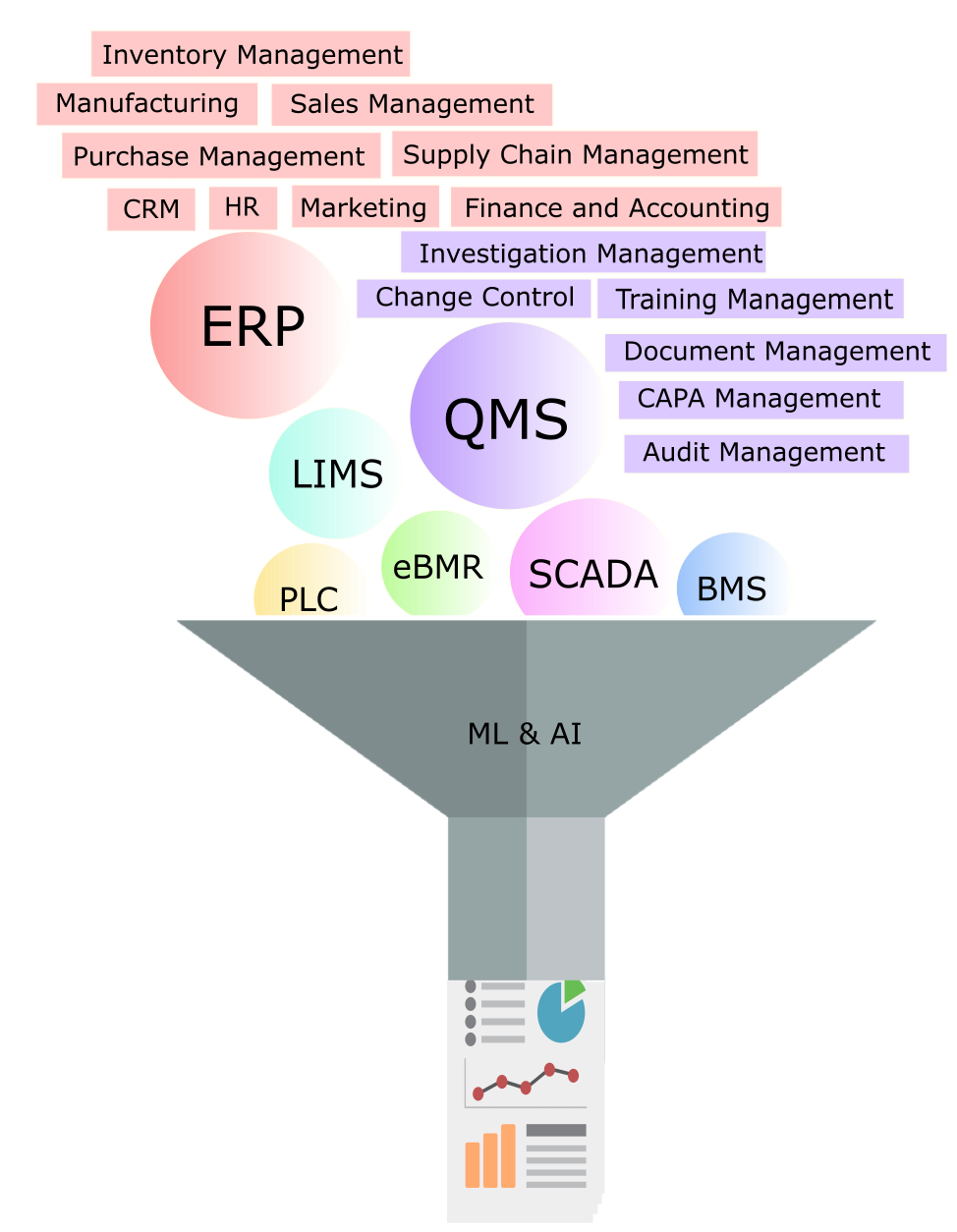
Figure 2: Integrating GxP and business systems for knowledge management
Integrate Systems To Meet Regulatory, Business Needs
To meet the regulatory need, integration of source data of material attributes, process parameters, and quality attributes is required. This is typically achieved by an electronic system that includes the enterprise resource planning (ERP) system, laboratory information management system (LIMS), quality management system (QMS), manufacturing equipment, and any other electronic data sources. Easy and quick access to the Stage 1 development data and integration of routinely used statistical assessment tools and risk assessment software are also critical to achieve the goal of implementing a seamless process validation life cycle management system per regulatory expectations.
The process validation life cycle management system (PVLMS) is expected to have the entirety of Stage 1, 2, and 3 data all in one easily accessible platform. The availability of data from all stages ensures knowledge management and use of data through effective search query functions. Integrated statistical analysis tools, risk assessment tools, and online Stage 3 (CPV) trending tools add tremendous value to any organization. Effective harnessing of Stage 3 (CPV) data is a pharmaceutical industry business requirement. The value proposition is realized in terms of reduced process failure rates, early identification of process losses, and even minimizing developmental studies with use of existing similar process data.

Figure 3: Minimizing development, documentation, and quality costs with use of integrated tools
FDA Warning Letters Indicate Lack Of Adoption Of CPV
Despite all these benefits, review of warning letter trends highlights the industry’s lack of well-developed process validation life cycle approaches. Specifically, deficient or nonexistent Stage 3 (CPV) programs contributed significantly to the issuance of FDA warning letters. The FDA has used similar language in warning letters and has recommended the use of a consultant, 5 indicating a lack of awareness/slow adoption of Stage 3 (CPV) by the industry. Manufacturing quality issues such as the proper execution of the process validation life cycle have been highlighted by the FDA as the biggest reason for drug shortages in the U.S. 6 The latest 2020 warning letter trends continue to indicate that there is an evident gap in understanding and adoption of the basic ideas. .
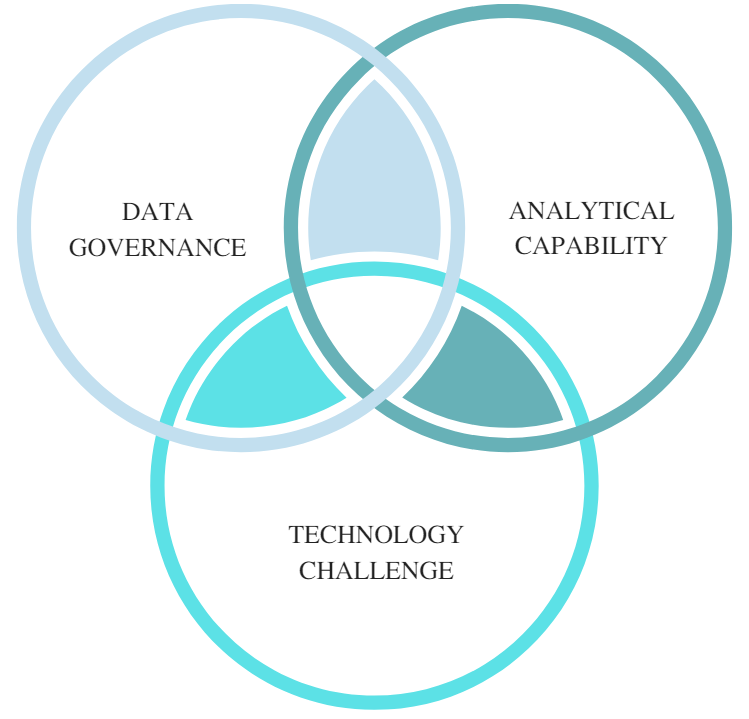
Figure 4: Factors deterring the adoption of CPV
When examining the reasons for the limited adoption of CPV, three factors stand out, with the first being lack of data governance. The high-volume manufacturers (generics representing almost 90 percent of prescriptions volumes), many of which are still correcting their basic data integrity issues, as continually highlighted by the regulatory bodies. Adequate data governance policies and controls are prerequisites for ensuring minimal integrity incidents. On the other hand, many innovative organizations are highly dependent on contract manufacturing organizations (CMO) for their product manufacturing needs. The use of multiple CMOs presents a challenging situation, as the maturity level, data access, integration, and governance differ from manufacturer to manufacturer.
The second deterrent is the lack of statistical/analytical experts with pharmaceutical science understanding, as well as the pharmaceutical scientists lacking daily use exposure and training on modern statistical tools. This often leads to using inappropriate data analysis methods that may not be applicable for the type of critical process parameter (CPP) or CQA signal observed. Finally, an all-encompassing (Stage 1, 2, 3) manufacturing network integrated process validation lifecycle management system (PVLMS) is still a distant reality. Recent stand-alone technology advancements in establishing digital facilities are a step forward. However, integration of process knowledge and use of manufacturing network technology interfaces continues to be limited at existing facilities. Application of process knowledge can improve operational capacity and process innovation throughput. There are plenty technology solutions available for each segment of the process validation life cycle and there are also highly effective stand-alone systems for statistical analysis, risk assessment, and statistical process control (SPC) trending. However, integrated solutions with effective knowledge management of Stage 1, 2, and 3 data are still evolving. Some new solutions that have been recently implemented are making great strides in holistically addressing process validation life cycle needs. Further optimization and applying machine learning and artificial intelligence (AI) solutions can augment organizational capabilities.
Conclusion
Enhanced product/process knowledge gained by the collection and evaluation of Stage 3 (CPV) data provides business value for manufacturers. A well-established Stage 3 (CPV) gathers the largest amount of data for the product, an organizational asset that leads to continuous improvement, reliable supply, product extensions, innovations, and intellectual property. Though the FDA’s process validation guidance has been in place for a long time and other regulatory agencies (EMA, WHO, PIC/S) continue to accept the Stage 3 (CPV) concept, adoption has been daunted by the data governance, analytical capability, and technology challenges. ICH Q12’s 7 recommended solutions such as the post approval change management protocol (PACMP) require extensive product/process data and continual trend analysis to ensure process control. Allowance for flexible post-approval changes with data will further boost the need for an all-encompassing effective product/process knowledge management solution, the process validation life cycle management system (PVLMS), in the pharmaceutical industry.
References:
About The Authors:

Ajay Pazhayattil is an industrial pharmacist with pharmaceutical operational experience in sterile, solid oral, and API sectors of the industry. He has been with North American brand, generic, and CDMO organizations in leadership roles prior to establishing cGMP World, an organization helping pharmaceutical firms maintain regulatory compliance. He has successfully remediated process validation warning letter scenarios at large generic organizations. He is a contributing author, speaker, committee lead and influencer in the industry and has been part of developing and delivering regulatory acceptable unique process validation solutions. He can be contacted through LinkedIn.

Marzena Ingram is an independent senior pharmaceutical consultant with extensive quality, technical operations experience in solid-dose and active pharmaceutical ingredients manufacturing operations. She is currently responsible for delivering compliant solutions for clients addressing FDA warning letter scenarios. Ingram has developed a specialized continued process verification team and spearheaded the implementation of the program to meet global regulatory requirements. She has also developed and published statistical tools for pharmaceutical manufacturing processes and has lead implementation of a comprehensive life cycle management software. She is a board member of ISPE Canada. She can be contacted through LinkedIn.

Eduardo Duyan is an accomplished pharmaceutical industry professional with expertise in multiple dosage forms including sterile injectable, transdermal, and solid and powder dosage forms. He has over 30 years of pharmaceutical and biotechnology industry experience. Duyan has helped pharmaceutical organizations address their consent decree, quality, and process remediation requirements. He has completed multiple overseas consulting assignments and is experienced in electronic data management systems including validation and configuration, data migration and management, and development of workflows. He can be contacted through LinkedIn.

Victor Zurita founded Compliance Associates Corporation in 2003 and serves as its president and CEO. He has held high-level positions in in the pharmaceutical industry in the areas of IT and GMP compliance. Zurita has built strategic partnerships with leading manufacturers and service providers to the life science industry including Thermos Fisher Scientific and IBM. With over 33 years of experience in the life science industry, he has lectured and provided training in North America and abroad on leading-edge topics in the compliance arena. Zurita possesses professional accreditation in the fields of chemical engineering and computer science. He can be contacted through LinkedIn.